Meet the CDG UK community
CDG UK is made up of our founders, trustees, advisory board, families and, of course, our generous donors.


Dr. Stephanie Grünewald
Patron & global CDG expert
We work closely with our patron, Dr Stephanie Grünewald of Great Ormond Street Hospital (GOSH), to distribute our information, and to keep updated on the latest developments in the world of CDG. She is an expert in this rare field and helps us to provide the information to the community of CDG families in the UK.
Dr Grünewald sees nationally and internationally referred CDG patients, coordinates diagnostics and research at GOSH and ICH, and collaborates with national and international clinical and scientific CDG networks.
Our advisory board
We are grateful to be guided by our advisory board, made up of experts in CDG and metabolic medicine, when making decisions with regards to supporting various new avenues of clinical research.

.png)

.jpg)






Meet our CDG heroes
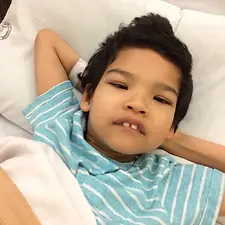
Lauren (30) has PMM2-CDG and didn't receive her diagnosis until the age of 25. In this interview, she tells us what that was like, and about the day-to-day experience of living with CDG as an adult.
.webp)
Finnan
At 5 months, when Finnan clearly wasn’t making any attempt to roll over, and didn’t seem to be able to see very far, we made an early appointment to see the Health Visitor for his 6 month review. From there, he was referred to the local hospital and then to Birmingham Children’s Hospital, where at 12 months he was diagnosed with CDG type 1a. Very soon after, his life became filled with appointments and therapists of all kinds. When he was three, he was lucky enough to get him a place in an outstanding local special school. Since then he has grown into the amazing little boy that we see today. He sits independently on a bench during circle time, he is able to move himself from lying down to sitting, and, most excitingly, he has just started walking at school with the aid of a walker. Finnan has also developed in leaps and bounds cognitively too. Recently, his vocabulary has developed very quickly and he has started to put words together to make meaning. He remembers stories from books and can count and add. He also thinks naughtiness is very funny and his best friends tend to be the naughty ones!

Ed
Our Edward is 9 years old. He had a natural birth at full term. During pregnancy, scans revealed that he had short long bones in his arms and legs. His head measurement was larger than expected and his stomach was distended. Ed’s hands were tightly clenched in a fist formation. We were told that there was a possibility that Edward had a chromosomal disease. Regular scans were made throughout pregnancy. Little boy lying down When he was born, Ed had a seizure and prolonged acute jaundice and spent the first two weeks in the Special Care Baby Unit at Winchester hospital. Once home, Edward was slow to feed and very floppy. He was never an active baby and it was clear that he was becoming unwell. At eight weeks of age, Edward was admitted to Southampton General Hospital with hypertension and acute renal failure. This was the beginning of a nine week stay in hospital, during which time he had peritoneal dialysis and emergency surgery for a perforated duodenal ulcer. Initially we were told “it does not look good” as Ed was so unwell. Surgery for the duodenal ulcer was successful, but with Ed’s kidney problems, we were told that Ed would probably never pee again. However, they had not reckoned his stubborn determination! Ed did start peeing again, but he had high levels of protein in his urine and was diagnosed with Congenital Nephrotic Syndrome (loss of protein through the kidneys). Ed had a kidney biopsy performed to try and find answers for his acute renal failure, but he suffered an internal bleed which required more surgery. He had an embolization to stop the bleed. He has 30% loss of function in his left kidney as a result. However, over time, Ed’s kidney function has slowly improved and at three years old, the Nephrotic Syndrome resolved itself! He now has a pretty normal kidney function. During his stay at Southampton General Hospital, Ed’s Renal Consultant, Dr Rodney Gilbert, suggested testing for CDG. He said that it was “a long shot” but he wanted to tick it off the list of possible causes for all of Ed’s problems. The results came back positive for CDG Type IIx. Since that time, a number of tests have been carried out to establish Ed’s sub-type. Sanford-Burnham Medical Research Institute in California discovered that Ed did not match any known sub-types, but has a mosaic mutation of this. Ed’s CDG is not hereditary, but a spontaneous mutation. He is SLC35A2-CDG. At the time, Ed was one of three children worldwide with SLC35A2-CDG and the results were written up in the American Journal of Human Genetics. Ed had severe reflux and was in a lot of pain and discomfort with it. It was also affecting his ability to gain weight. A couple of years ago Ed had a fundoplication, via keyhole surgery, to stop his gastroesophageal reflux. This has helped Ed to gain weight. Ed still has poor muscle tone, but has a great deal of determination. It has taken him a while, but Ed can now hold his head for a short while and has mastered rolling over. He is trying to crawl by pushing his knees forwards, but he does not have the upper body strength to weight bear with his arms. Ed is tube fed and only eats a small amount of solids a day. Ed cannot speak but he does vocalise and is trying hard to make words. He can say “Edward”, “mummy”, “brother”, “Hello”, “yeah” and “babby” (daddy). We and the school believe that Ed will one day speak a few words! Ed also loves to blow raspberries!! Ed started school just before his fourth birthday. He settled in straightway and particularly enjoys hydrotherapy, sensory play and tactile music activities. Ed is a happy smiley and very determined little boy. Everyone who meets our happy, smiley little boy falls in love with him.

Ruby
gained her wings Jan. 2026
Ruby was born at 42 weeks and discharged from hospital the very next day. At around 3 weeks she was struggling with feeding and weight gain, and then at 8 weeks she began presenting with numerous unexplained symptoms, such as a mass of fluid in her belly (ascites) and around her heart (a pericardial effusion). At 3 months of age she was diagnosed with PMM2-CDG, and from that point spent most of her life in hospital, until, very sadly Ruby passed away in January 2026. Despite her many challenges, Ruby remained a happy and resilient little girl, who managed her way through a liver transplant before she had even turned two, with her mum Elle being her donor. Ruby showed incredible strides in her development and met many milestones that her parents didn't necessarily expect to see her achieve so soon. She loved music, school and was a true sensory explorer. Ruby's presentation of PMM2-CDG was so complex and severe, that her doctors learned a lot of new things from her short life.

Thomas
Asking our GP to look at Tom's squint and floppiness, when he was 7 months young, put us on the rollercoaster journey to the diagnosis of PMM2-CDG at 2 years old. Between uninformed medical professionals and unhelpful google resources, we had been prepared for the worst. Thankfully, 10 years later we have a vibrant boy who has a wonderful zest for life, always curious and wanting his own way of course. He is a caring brother with a big heart, a cheeky smile, and amazing determination. Thomas has come to far thanks to the early intervention of all the therapies, being able to take part in an extended clinical trial, and having the right support from our CDG experts, including CDG UK, to whom, as a family we will forever be grateful to!

Matthew
gained his wings Dec. 2019
Matthew was born at home following an uneventful pregnancy and after a 45min labour, he came out feet first (we didn’t know he was footling breech) Surprise! The first 12 days went fine except he would twitch a bit when falling asleep. Numerous midwife and health visitor visits reassured me it was nothing serious although when we were admitted to hospital due to losing 10% of his birth weight, on the ward I showed the doctor the twitching and she confirmed they were seizures. An urgent MRI, Lumbar puncture, ng tube and antibiotics were done, luckily confirming it wasn’t the meningitis they suspected but sadly confirming Matthew had two bleeds on the brain, they expect from his quick and upside down birth. We left the ward a week later being told a neurologist would tell us more but they couldn’t say if he would be a average child and he would possibly be delayed. After 3 further months of fighting to get him to feed, he was diagnosed malnourished and fitted long term with an ng tube and we saw the neurologist who blew us away with the fact the bleeds were not the cause of Matthews issues of floppiness, poor feeding, poor vision, poor hearing, poor social interaction but that he thought it was genetic. He noted inverted nipples, poor fat distribution, cross eyes and sent some blood off for testing. When Matthew was 6 months old we went to sign some paperwork to start a CAF (basically a meeting to get all professionals together) and was hit with the bombshell that they had diagnosed Matthew with Congenital Disorder of Glycosylation. Type unknown. Expected 1A with his symptoms. To be honest the diagnosis was a relief to know what had happened the hardest bit was finding out it’s a 1 in 3 chance to happen every pregnancy. In the next six months we found he had a heart murmur, fat spheres on his kidneys, his liver function is abnormal and he has optic nerve hypoplasia (poor number of nerves), he has poor muscle tone everywhere and I mean everywhere. Matthew is now almost 3, about 2 years ago he had a skin biopsy confirming he is type 1A, he still cannot sit or stand. He cant grab a toy or hug you. He cant say words but can say mama and dada although its more of a ma ma ma ma :). He is now fed through a tube in his stomach firstly with a PEG now a Mini Button. Although he is now taking some fruit puree but this has been a long road. He’s a typical toddler obsessed with the Ipad. He’s a social little bunny and has an incredible smile. He has just started school and seems to be thriving there. We are still at a very early road with him and we thank whoever is upstairs for every year we have with him.
.jpg)
Rosie
Meet Rosie. Rosie is 11 years old and loves to sing, dance and play with her friends. Rosie also has a DHDDS genetic mutation which causes both a CDG and lysosomal storage disorder. Rosie is full of fun, life and laughter but also battles daily with tremors, balance and co-ordination difficulties and learning difficulties. Our hope is that we can find a treatment for Rosie so that she can have a brighter future and keep doing the things she loves.

Emily
Becky and Phil Hamilton of Accessible Holiday Escapes got in touch with CDG UK to share their family’s story. Emily was diagnosed with CDG Type 2 in 2015 at the age of 3. She was born at full-term and everything progressed as expected at first. When Emily was about 7 or 8 months old we began to get more concerned about her physical progress as she wasn’t sitting up, rolling or developing any fine motor skills. Other than that she seemed very healthy and happy. Emily was referred to the community paediatrician at around 12 months old, and since then physiotherapy, speech therapy, occupational therapy and much more has been ongoing. Emily underwent investigations in the form of blood tests, scans, and assessments by different specialists at various hospitals. Ataxic Cerebral Palsy was initially suspected. Emily standing with frameEmily sat up at 14 months old, crawled at 18 months old, walked using a rollator aged 2, and walked independently at around 2 and a half. For a long time she needed lots of help and support when walking because of her poor balance and still needs help now. although is doing far better than we ever hoped! Emily is very sociable and happy, and also very determined and stubborn! Emily has learning disabilities, and spent 4 years in our local mainstream school with 1:1 support, and has just moved to a resourced provision within a mainstream school with a specialist teacher and staff. In 2018 we began writing a review-style family accessible adventures blog (we also have an 11 year old son Sam) www.theflyingbumblebee.com to share our days out/holiday experiences and became part of the large disability community online. We then realised it wasn’t just us who struggled with finding accessible family holidays. As Emily became older and our needs became more specific, we found thatthere seemed to be very little information or search options on-line for accessible and child-friendly properties, and it was difficult to find the access information we needed. As a family of 4 it’s really important to us to find somewhere that will be right for all of us, but unexpected internal steps, missing stair gates, or open gardens mean it suddenly isn’t a holiday any more! Emily on a horseWe eventually decided to take the plunge and launch our own website. The accessible tourism industry is growing in the UK so we felt it may also be successful as a business, as well as helping travellers like us search for accessible holidays. We launched www.accessibleholidayescapes.co.uk in June 2019 and have had an amazing response. We already have 70 properties listing with us and more due to be uploaded; and have had a really positive response from users of the site. We won a Theo Paphitis Small Business Sunday award recently, and were filmed by ITV Wales news talking about our story and the website, and were featured in the Daily Post newspaper. We also hope that sharing our story will raise awareness of CDG. We have yet to meet a professional (other than the team at BCH) who have worked with a child with CDG before, so Emily is usually their little guinea pig! Our unexpected CDG journey has eventually led us down the path of accessible tourism and given us a totally different outlook on life. Like any parents of a child with a disability some days can be difficult, and the appointments, paperwork and therapy programmes are never-ending; but we try and maintain some sort of happy family life with accessible adventures thrown in!
.jpg)
Malakai
gained his wings May 2024
On the 31st of March 2024, at 11:11pm, our beautiful baby boy Malakai was born via emergency ceserean. To us, his parents, he was perfect. But a Dr in the NICU made a passing comment about how they think that there is something genetically going on because of the bridge of his nose being a little wide. I was induced 24 hours prior to the ceserean, because Malakai had reduced movements quite often. Which we now know was most likely caused by his low muscle tone. He was with us on the ward for a few hours, whilst I was trying to harvest colostrum. After he had some colostrum, I noticed a very slight gargling sound when he was breathing, like he had mucus stuck in his throat. I flagged it up with the midwife but they dismissed it initially, when I mentioned it again they had someone come and take him to have a suction tube clear his airways. The noise had gone, but then came back after he had more colostrum. At this point I started to get really worried and still felt like I was being dismissed. They offered to suction his airways again, but another lady came and took his blood sugar. This is when he was rushed to the NICU because his levels were dangerously low. At this point, my legs were still numb so I couldn’t go with him. But my partner/Malakai’s dad, was able to go so he wasn’t alone. This was the beginning of a very long and painful 6 weeks. So all we knew was that Malakai was unable to keep his milk down. We were there for approximately a week and a half, Malakai was having numerous tests. Malakai had ultrasounds to check his organs and a 24 hour brain scan to monitor his brain activity. This was to check for any seizures. These tests all came back normal, apart from his heart having 3 holes, but we were told this is normal for babies and that they would close up over time. We were left confused, with no answers as to why he couldn’t keep his milk down. Me and my partner brought it up with the Dr that perhaps we could be transferred to St Thomas’s hospital as they had more experienced staff and better equipment. They initially said no, but later realised something wasn’t right and transferred us a day or two later. The first couple of days we were at St Thomas’s, Malakai underwent an MRI on his brain. Which we got the results for a few days later maybe. Malakai’s Cerebellum and Pons were under-developed, and we still didn’t know what this meant. But we were ready to take on what challenges this would have brought. After more tests, numerous cannulas inserted, NG, NJ tubes and internal lines fitted, they decided we needed to have blood taken from us so that the genetics lab could compare it to Malakai’s blood to check for genetic conditions. From what I remember, this took approximately three weeks. We were left in a state of confusion, anger, and sadness. I was trying to express milk because it was the only thing I could do, but I had a low supply due to my Polycystic Ovary Syndrome. Having to walk away from our baby every day, so that we could sleep and eat, was extremely difficult and we felt (and still do) a lot of guilt. The time had come for the professor to give us the results. We were led to an empty room, with a circle of chairs, to fill staff members from different areas of the unit. The geneticist told us that they found a genetic condition, and that it affected a very low number of babies worldwide. I asked what does all of this mean? He said, it means Malakai has a life expectancy of 6 to 12 months. And just like that, mine and my partner’s life absolutely shattered into a million pieces. The geneticist explained that me and my partner are both carriers for the CDG gene, but my partner’s gene has ‘pages missing’ and mine has ‘spelling mistakes’. This meant Malakai has ALG1-CDG, a rare form, which he had a 1 in 4 chance of inheriting. Now everything started to make sense. Malakai had low muscle tone in his chest, meaning he couldn’t keep milk down. The bridge of his nose the Dr spoke about, his legs and arms being slightly stiff. These were all such subtle signs. During my pregnancy with Malakai, there were no red flags apart from having one vessel in the umbilical cord, his legs were measuring shorter than average and my alpha protein levels were slightly abnormal. We were referred to the Harris Birthwright centre to regularly see a fetal medicine specialist for regular scans. They were happy with how things were going, apart from his leg measurements. We spent two nights in the hospital with Malakai, before being discharged on the 8th of May so that we could take him home to pass away comfortably. Palliative care is not something we thought we would ever hear when it came to our baby. We had a visit from a lady who worked at our local Demelza Hospice. We didn’t realise that we had the option of being there and cared for. We stayed at home for two nights, so that Malakai could use all the things he wasn’t able to due to being in hospital. We also took him for a short walk in his pram. But in the early afternoon of the 10th of May, after a visit from the palliative care team and a few immediate family members, I think we knew that the time was coming. We arrived at Demelza a few hours later, and were welcomed by lovely, caring, happy staff members. The staff helped us to make some memories with clay casts, and a few more family members dropped by to say goodbye. And before we knew it, at approximately 8pm our baby boy took his last breath. The night Malakai passed away, northern lights appeared outside, which I believe was his way of telling us he was okay now. We spent 5 days with him at Demelza, and we are incredibly grateful for what they have done for us.